Diseases
Click here to view more Diseases

Wilson's disease (WD), also known as hepatolenticular degeneration, is an autosomal recessive disorder characterized by impaired hepatic copper transport and subsequent copper accumulation in various tissues, particularly the liver, and brain.
The exact prevalence of Wilson's disease in India remains uncertain due to underdiagnosis and a lack of population-based studies. However, several studies indicate that India has a higher prevalence of WD compared to Western countries. The high frequency of consanguineous marriages and the increased occurrence of WD in certain communities contribute to the disease burden in India. A study conducted by Shalimar et al. in 2019 reported the prevalence of Wilson's disease to be approximately 7.4 per million population in northern India, which is higher than previously estimated. Another study by Sood et al. in 2020 observed a higher prevalence of Wilson's disease (16.6 per million) in the northern Indian state of Punjab. These findings highlight the need for further research and comprehensive nationwide studies to ascertain the true prevalence of Wilson's disease across different regions in India.
Wilson's disease is characterized by impaired copper transport and metabolism in the body. the complex biochemical pathways and pathophysiology of Wilson's disease are illustrated below :
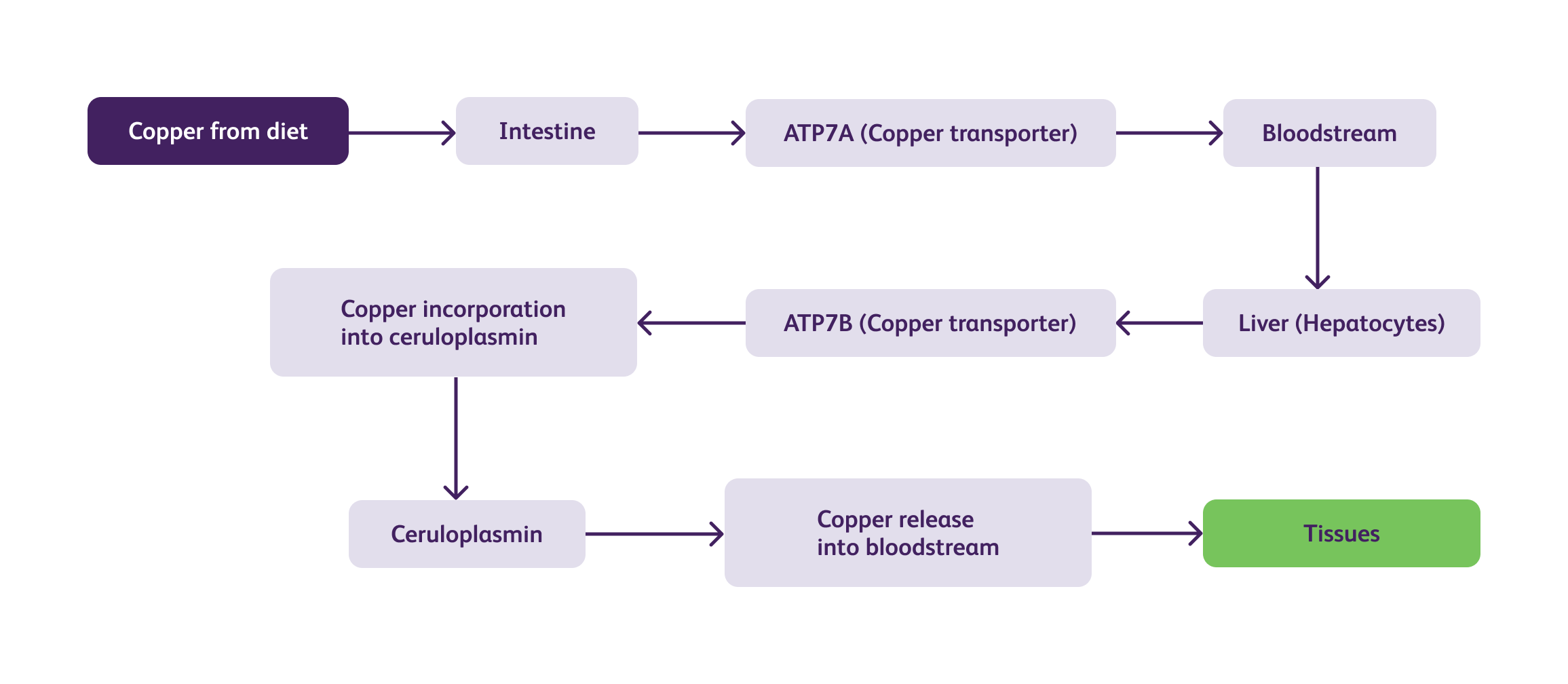
In Wilson's disease, mutations in the ATP7B gene impair the function of the ATPase copper-transporting beta (ATP7B) protein, leading to defective copper transport. As a result, copper accumulates within the liver and is not adequately incorporated into ceruloplasmin, resulting in low levels of circulating ceruloplasmin. The excess copper released into the bloodstream is then deposited in various tissues, including the liver, brain, kidney, and cornea, causing tissue damage and dysfunction.
The clinical manifestations of Wilson's disease are highly variable and can involve multiple systems.
The management of Wilson's disease involves two primary goals: reducing copper accumulation and preventing its toxic effects.
Enhances urinary copper excretion by forming a complex with copper.
Similar to D-penicillamine, it enhances urinary copper excretion but with
fewer side effects than D-penicillamine and is advised in neuro-Wilson’s disease.
(For
more information click here)
Considered in cases of severe liver damage or failure that do not respond to medical treatment.
| Aspect | D-penicillamine | Trientine Tetrahydrochloride |
|---|---|---|
| Dose | Initial: 250 mg/day | Initial: 750-1,500 mg/day in divided doses |
| Gradually increased to 1-1.5 g/day | Gradually increased to 750-2,250 mg/day (divided into 2-3 doses) depends on patient's response | |
| Contraindications | Hypersensitivity to penicillamine | Hypersensitivity to trientine |
| Pregnancy and breastfeeding | Pregnancy and breastfeeding | |
| Neurological Deterioration | Has been reported, and treatment should be promptly stopped if any signs of worsening neurological symptoms occur | Not reported |
| Adverse Effects |
- Rash, fever, proteinuria - Hematological abnormalities - Nephrotoxicity - Autoimmune disorders - Neurological symptoms |
- Gastrointestinal disturbances - Neurological symptoms (rare) - Hematological abnormalities - Nephrotoxicity - Autoimmune disorders - Neurological symptoms - Dermatological reactions - Myasthenia gravis (rare) |
| Contraindicated drugs/Drug-Drug Interactions |
Gold compounds Antimalarials Cytotoxic drugs Aluminum-containing drugs Iron salts Magnesium salts Zinc salts Calcium salts Nonsteroidal anti-inflammatory drugs (NSAIDs) Tetracyclines Captopril Nitrofurantoin Methyldopa Anticoagulants Digitalis glycosides Meclofenamate Sulfinpyrazone Oral penicillins Oral cephalosporins |
Aluminum-containing drugs Iron salts Magnesium salts *Zinc salts Calcium salts Nonsteroidal anti-inflammatory drugs (NSAIDs) Captopril Nitrofurantoin Methyldopa Anticoagulants Digitalis glycosides Meclofenamate Sulfinpyrazone penicillins Oral cephalosporins |
*Zinc supplements: Zinc supplements may decrease the absorption of Trientine. These medications should be administered at least two hours apart. (Reference: BNF, Trientine Tetrahydrochloride monograph).
✓ Low Copper Diet: Restricting dietary intake of copper-rich foods such as liver, shellfish, mushrooms, and chocolate.
Several innovative approaches have emerged for the management of Wilson's disease. Novel copper chelators, such as tetrathiomolybdate, have shown promising results in reducing copper overload. Gene therapy and RNA interference techniques hold potential for targeted gene correction and silencing, respectively. Additionally, advancements in imaging techniques, such as molecular imaging, may aid in early diagnosis and monitoring of treatment response.
Wilson's disease typically manifests between the ages of 5 and 35, with most cases presenting in adolescence or early adulthood. In India, the age of onset appears to be slightly earlier compared to Western populations, with a peak incidence in the second decade of life. This younger age of presentation poses unique challenges in terms of diagnosis and management, as symptoms can be easily mistaken for other common conditions in this age group. Despite advancements, challenges persist in managing and diagnosing Wilson's disease in India. Limited awareness among healthcare professionals and the general population often leads to delayed or missed diagnoses. Inadequate access to specialized diagnostic facilities and genetic testing further hinders accurate identification of the disease. Long-term treatment adherence and monitoring pose additional challenges, necessitating comprehensive multidisciplinary care.
Future research in Wilson's disease should focus on improving diagnostic algorithms, particularly in resource-limited settings. Efforts to raise awareness among healthcare professionals and the public are crucial for early detection and intervention. Collaboration between clinicians, researchers, and policy-makers is necessary to establish national registries, facilitate genetic testing, and develop tailored management guidelines. Continued exploration of targeted therapies and gene-based interventions holds promise for personalized treatment approaches.

Disclaimer
 Healthcare Professional
Healthcare Professional